Спинальная мышечная атрофия
и Флориан Тифенбек, врач ОбновлениеМаксимилиан Рейндл изучал химию и биохимию в LMU в Мюнхене и является членом редакционной группы с декабря 2020 года. Он ознакомится с медицинскими, научными и политическими темами для вас, чтобы сделать их понятными и понятными.
Другие сообщения Максимилиана РейндлаФлориан Тифенбек изучал медицину человека в LMU в Мюнхене. Он присоединился к в качестве студента в марте 2014 года и с тех пор поддерживает редакцию медицинскими статьями.После получения медицинской лицензии и практической работы по внутренним болезням в Университетской клинике Аугсбурга он с декабря 2019 года является постоянным членом команды и, помимо прочего, обеспечивает медицинское качество инструментов
Другие сообщения Флориана Тифенбёка Весь контент проверяется медицинскими журналистами.
Спинальная мышечная атрофия, или сокращенно СМА, - это редкое заболевание, при котором умирают определенные нервные клетки спинного мозга. Стимулы и импульсы из мозга больше не достигают своего пункта назначения: мышц. Это вызывает мышечное истощение и паралич. Существуют разные формы SMA. Самое тяжелое начинается в младенчестве. Новые методы лечения обещают долгосрочное улучшение здоровья. Подробнее о мышечной атрофии позвоночника читайте здесь.
Коды МКБ для этого заболевания: коды МКБ - это международно признанные коды медицинских диагнозов. Их можно найти, например, в письмах от врачей или в справках о нетрудоспособности. G12

Краткий обзор
- Что такое спинальная мышечная атрофия? Группа заболеваний мышечной слабости. Они основаны на гибели определенных нервных клеток спинного мозга, которые контролируют мышцы (мотонейроны). Следовательно, SMA относятся к числу заболеваний двигательных нейронов.
- Какие бывают формы? Наследственные спинальные мышечные атрофии - это в основном СМА с определенным генетическим дефектом на хромосоме 5 (5q-ассоциированная СМА). Врачи различают четыре разные формы: СМА типа 1 - типа 4. Кроме того, существуют спорадические формы, наследственность которых не установлена.
- Частота: редкое заболевание; наследственная СМА поражает примерно одного новорожденного из 7000.
- Симптомы: подергивание мышц, прогрессирующая мышечная слабость, мышечное истощение, паралич. Градиенты различаются в зависимости от формы SMA.
- Причины: наследственная спинальная мышечная атрофия 1-4 типов является результатом генетического дефекта хромосомы 5, а точнее гена SMN1. В результате в организме отсутствует особый белок - белок SMN. Этот дефицит повреждает мотонейроны спинного мозга.
- Диагностика: генетическое обследование на предмет изменения генетической структуры SMN, физикальное обследование, электронейрография, электромиография, анализы крови (например, CK)
- Лечение: возможна заместительная генная терапия или введение модуляторов сплайсинга. Сопутствующая физиотерапия, логопедия, обезболивание и психотерапия. При необходимости операция на позвоночнике. План лечения в зависимости от типа СМА.
- Прогноз: В случае наследственной проксимальной СМА новые варианты терапии имеют причинный эффект и могут положительно повлиять на течение болезни. Раннее начало лечения имеет решающее значение. Лечение доступно не каждому пациенту. Если не лечить, дети со СМА типа 1 обычно умирают в течение первых двух лет. Продолжительность жизни у 3-го и 4-го типа практически не снижается.
Что такое спинальная мышечная атрофия?
При спинальной мышечной атрофии (СМА) некоторые нервные клетки спинного мозга умирают. Обычно они контролируют мышцы, поэтому специалисты называют эти нервные клетки двигательными нейронами. Соответственно, СМА относятся к так называемым заболеваниям двигательных нейронов.
При спинномозговой мышечной атрофии поражаются нижние (вторые) мотонейроны, напрямую связанные с мышцами своими придатками. В результате повреждения меньшее количество нервных сигналов или их полное отсутствие достигают мышц. Мышцы становятся все слабее и меньше (истощение мышц / атрофия мышц).
Врачи различают разные формы мышечной атрофии позвоночника. Безусловно, самая большая группа - это наследственная СМА, при которой поражаются мышцы, близкие к туловищу (проксимальные). В их основе лежит конкретный генетический дефект. Он разовьется примерно у одного из 7000 новорожденных.
Спинальная мышечная атрофия - в целом редкое заболевание. Тем не менее, это второе по распространенности аутосомно-рецессивное наследственное заболевание. Это также самая частая причина смерти младенцев или детей ясельного возраста из-за генетического дефекта.
Какие бывают типы мышечной атрофии позвоночника?
Врачи дифференцируют наследственные (наследственные) формы СМА от спорадических. Другая классификация спинальной мышечной атрофии относится в первую очередь к группам мышц, пораженным в первую очередь. Есть
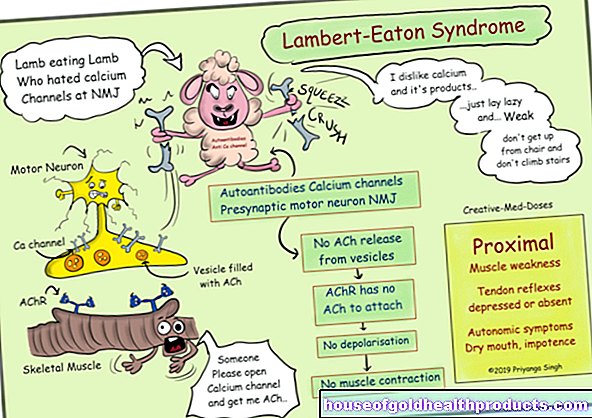
- Проксимальный SMA: около 90 процентов, они составляют самую большую группу SMA. Симптомы начинаются в мышцах около туловища, то есть проксимально.
- Непроксимальная SMA: здесь в первую очередь поражаются более отдаленные группы мышц, такие как руки и ноги (дистальная SMA). В дальнейшем эти SMA могут также распространяться на мышцы около середины тела.
- Особые формы (например, спинобульбарная мышечная атрофия типа Кеннеди)
Проксимальные мышечные атрофии позвоночника
Наследственные проксимальные мышечные атрофии позвоночника - это, в основном, заболевания, в основе которых лежит определенный генетический дефект (5q-ассоциированная СМА, дефект на хромосоме 5). Они, в свою очередь, делятся на четыре разные формы. Классификация основана на времени появления первых симптомов и течении болезни.
Спинальная мышечная атрофия 1-го типа: это наиболее распространенная и наиболее серьезная форма СМА. Ее также называют «болезнью Верднига-Гофмана» или «острой инфантильной СМА». Заболевание обычно начинается в раннем детстве. Слабость мышц влияет на все тело - врачи также говорят о «синдроме гибкого младенца» (от английского floppy = вялый, младенец = младенец, ребенок). Большинство нелеченных детей с СМА типа 1 умирают в возрасте до двух лет.
Спинальная мышечная атрофия 2 типа: эта форма СМА также известна как «промежуточная спинальная мышечная атрофия» или «хроническая инфантильная СМА». Первые симптомы обычно появляются в возрасте до 18 месяцев. У пострадавших иногда значительно сокращается продолжительность жизни.
Спинальная мышечная атрофия 3 типа: она также известна как «ювенильная спинальная мышечная атрофия» или «болезнь Кугельберга-Веландера». Этот СМА обычно начинается в возрасте после 18 месяцев и до раннего взросления. Слабость мускулов более легкая, чем при типе 1 или 2. Ожидаемая продолжительность жизни у пострадавших лишь немного сокращается.
Спинальная мышечная атрофия 4-го типа: похожа на СМА 3-го типа, но появляется только в зрелом возрасте (обычно> 30 лет). Однако мышечная слабость менее выражена и прогрессирует медленнее, чем при СМА 3 типа.
Переходы между различными версиями плавные. В некоторых случаях это затрудняет четкое разграничение. Некоторые генетические предрасположенности также играют важную роль в тяжести рассматриваемого заболевания.
Другие спинальные мышечные атрофии
Помимо этих проксимальных форм, существуют другие формы мышечной атрофии позвоночника. К ним относятся, например, более редкие наследственные дистальные мышечные атрофии позвоночника. У них симптомы обычно начинаются в группах мышц, находящихся дальше от тела.
В случае спорадического SMA наследование не гарантируется. Кроме того, невозможно определить семейное накопление. В литературе к ним относятся:
- Тип Хираямы (ювенильная дистальная СМА, заболевание в возрасте около 15 лет, поражает мышцы рук, обычно останавливается даже без терапии и может даже улучшиться)
- Тип Вульпиана-Бернхарда (также синдром «цепной руки» с началом в плечевом поясе, обычно с 40 лет)
- Тип Дюшенна-Арана (первоначально поражаются мышцы рук, распространяющиеся по направлению к туловищу, обычно после 30 лет)
- Малоберцовый тип (синдром «цеп-ножка», сначала на мышцах голени)
- Прогрессирующий бульбарный паралич (нарушение речи и глотания, поражает около 20 процентов пациентов с боковым амиотрофическим склерозом)
Некоторые спорадические формы СМА (синдром «цепной руки - / - ноги», прогрессирующий бульбарный паралич) считаются в кругах специалистов одними из вариантов бокового амиотрофического склероза (БАС). В данной статье рассматриваются в первую очередь наследственные проксимальные мышечные атрофии позвоночника.
Спинобульбарная мышечная атрофия
Спинобульбарная или бульбоспинальная мышечная атрофия (тип Кеннеди, синдром Кеннеди) - наследственное заболевание. Это часто начинается в молодом и среднем возрасте. Эта особая форма СМА наследуется рецессивным образом, сцепленным с Х-хромосомой, и поэтому поражает только мужчин (поскольку у мужчин только одна Х-хромосома, у женщин преобладает вторая, здоровая Х-хромосома, которая компенсирует дефект).
Часто встречается мышечная слабость в мышцах, прилегающих к телу, на ногах, руках или плечах, а также в мышцах языка и горла. В результате у пострадавших, например, возникают проблемы с речью и глотанием. Также они жалуются на тремор, мышечные спазмы и подергивания. Больные мужчины также часто имеют задержку роста яичек и бесплодие. Кроме того, увеличиваются молочные железы (гинекомастия).
Спинобульбарная мышечная атрофия обычно протекает медленно. Продолжительность жизни практически не ограничена.
Как распознать мышечную атрофию позвоночника?
Типичными для спинальной мышечной атрофии являются прогрессирующая мышечная слабость вплоть до паралича (пареза) и подергивания мышц. В результате повреждения нерва мышцы больше не получают электрические импульсы, что со временем приводит к их сокращению (атрофия мышц). Точные признаки и жалобы зависят от соответствующей формы. В следующем разделе рассматриваются симптомы наследственной проксимальной СМА.
Симптомы детской спинальной мышечной атрофии 1 типа
Симптомы СМА 1 типа появляются в первые шесть месяцев жизни. Возникает общая мышечная слабость, то есть такая, которая затрагивает все тело. Кроме того, уменьшается напряжение между мышцами. Врачи говорят о гипотонии.
У новорожденных эта мышечная слабость изначально проявляется в типичной позе ног, напоминающей лежачую лягушку (поза лягушачьей ноги). Ноги согнуты, колени развернуты наружу, а ступни - внутрь. Даже поднять или удержать голову самостоятельно обычно невозможно.
В преклонном возрасте дети с СМА типа 1 не могут самостоятельно сидеть или ходить. Многие дети также не могут говорить, так как могут быть затронуты мышцы языка.
Другой характеристикой спинальной мышечной атрофии 1 типа является форма верхней части тела: мышцы груди и спины не развиваются должным образом. Это придает верхней части тела колоколообразную форму (сундук с колоколом). Из-за слабого развития мышц груди и спины пострадавшие принимают сутулую осанку.
Часто также наблюдается увеличение искривления позвоночника (сколиоз). Сгорбленная поза и сгорбленная поза вызывают дальнейшие проблемы с дыханием. Характерно очень быстрое и поверхностное дыхание (тахипноэ).
Симптомы промежуточной мышечной атрофии позвоночника 2 типа
Спинальная мышечная атрофия 2 типа обычно вызывает симптомы только в возрасте от семи до 18 месяцев. Больные дети могут сидеть самостоятельно, но обычно не учатся ни стоять, ни ходить. Слабость мышц прогрессирует медленнее, чем при 1-м типе.
При СМА 2 типа со временем возникают симптомы, похожие на симптомы тяжелой детской формы, такие как деформация позвоночника. Суставы становятся жесткими из-за укорочения мышц и сухожилий (контрактуры). Другие признаки включают тремор в руках и подергивание мышц языка.
Симптомы ювенильной спинальной мышечной атрофии 3 типа
Спинальная мышечная атрофия 3 типа обычно появляется в возрасте от 18 месяцев до 18 лет. Больные дети могут сидеть, стоять и ходить самостоятельно. Однако слабость мышц, особенно мышц таза и ног, вызывает походку вразвалку.
В течение нескольких лет работоспособность снижается: сначала людям трудно заниматься спортом или подниматься по лестнице, но в конце концов, например, также трудно нести сумки для покупок. Спустя много лет мышечная атрофия позвоночника 3-го типа делает бег и любые другие нагрузки трудными или даже невозможными даже для пожилых людей.
Однако в целом симптомы менее выражены, чем при двух других формах заболевания, типе 1 и типе 2. Для многих из них качество жизни практически не ухудшается в течение длительного периода времени.
Симптомы спинальной мышечной атрофии 4 типа у взрослых
Эта очень редкая форма прогрессирующего мышечного истощения начинается в зрелом возрасте, часто с третьего десятилетия жизни. Первоначально поражаются мышцы ног и бедер. По мере прогрессирования болезни мышечная слабость распространяется также на плечи и руки.
Проявление клинической картины аналогично ювенильной СМА 3 типа. Однако прогрессирующая мышечная слабость происходит даже медленнее, чем при СМА 3 типа.
Что вызывает спинальную мышечную атрофию?
При спинальной мышечной атрофии вторые мотонейроны спинного мозга погибают. Это нервные клетки, которые контролируют мышцы своими придатками. В результате повреждения этих узкоспециализированных мотонейронов к мышцам поступает меньше электрических сигналов, чем у здоровых людей. Если мышечные клетки используются меньше и, следовательно, меньше стимулируются, организм со временем разрушает их.
Генетический дефект
В большинстве случаев спинальная мышечная атрофия является наследственным заболеванием (наследственная СМА). Причина типичных проксимальных форм СМА - неверная информация о генетическом составе пациента. Так называемый ген SMN1 на хромосоме 5 не работает.
Ген SMN1 несет информацию - то есть план - жизненно важной белковой молекулы, называемой SMN. SMN расшифровывается как «Выживание моторного нейрона». Без молекулы белка SMN мотонейроны со временем погибают.
Верно, что в организме также есть родственный ген SMN2, который в принципе способен «компенсировать» нефункциональную генетическую информацию SMN1.Но обычно это случается лишь в небольшой степени. Это означает, что потеря функции гена SMN1 (если ее не лечить) обычно не может быть полностью компенсирована интактной копией гена SMN2.
Аутосомно-рецессивное и аутосомно-доминантное наследование
Генетическая информация человека доступна в двух экземплярах. В результате у каждого есть две копии гена SMN1 - одна от отца и одна от матери. Проксимальные мышечные атрофии позвоночника в детском возрасте обычно наследуются как аутосомно-рецессивный признак.
Это означает, что оба варианта гена (аллели) от родителей должны быть дефектными для развития спинномозговой мышечной атрофии у потомства. В случае рецессивного наследования родители не страдают, потому что, помимо безфункционального, у них также есть здоровый ген SMN1, который компенсирует дефект.
Примерно каждый 45-й человек является обладателем этой системы для SMA. Пара, в которой оба партнера являются носителями, имеет 25% -ный риск рождения ребенка с этим заболеванием.
В некоторых случаях в подростковом возрасте спинальные мышечные атрофии, в частности, во взрослом возрасте, также возникают по аутосомно-доминантному наследованию. В случае доминантного наследования дефектный ген уже заявляет о себе - и пострадавшие заболевают. Однако это не вышеупомянутый генетический дефект на хромосоме 5. Эти 5q-ассоциированные SMA всегда наследуются аутосомно-рецессивным образом.
Наследование с другими формами SMA
Непроксимальная спинальная мышечная атрофия также может передаваться по наследству. Особая спинобульбарная форма (тип Кеннеди) наследуется рецессивно через половую хромосому, Х-хромосому (это влияет на варианты генов, которые содержат план стыковочных сайтов для мужских половых гормонов). Однако в случае спорадических форм наследование не гарантируется. Здесь мало что известно о причинах гибели вторых мотонейронов.
Обследования и диагностика
Диагноз спинальной мышечной атрофии обычно ставят педиатры, педиатры, специализирующиеся на нервных заболеваниях (нейропедиатры), и специалисты по заболеваниям нервной системы (неврологи). Для более точного выяснения необходимы различные исследования. В случае СМА особенно важны генетические тесты и исследования нервов и мышц.
Сбор анамнеза (анамнеза)
При каждом заболевании врач в первую очередь спрашивает о появившихся симптомах и о том, как оно прогрессировало. У младенцев и малышей родители сообщают об изменениях и отклонениях в поведении своего ребенка. В частности, в случае наследственных заболеваний врачи также обращают внимание на историю болезни семьи.
Физические осмотры
В основном, отклонения в двигательном развитии врач определяет, осматривая ребенка физически. Например, он проверяет, могут ли дети самостоятельно держать голову в вертикальном положении, сидеть или двигать руками или ногами независимо (в зависимости от их возраста).
Дополнительные тесты с физической нагрузкой проводятся у детей старшего возраста и взрослых с подозрением на мышечную атрофию позвоночника. Врач проверяет, сколько силы может набраться заинтересованного человека и как долго он сможет ее удержать. Он также проверяет выносливость.
Кроме того, врач проверяет рефлексы, которые обычно ослабляются или угасают, особенно в случае выраженных мышечных атрофий позвоночника. Для этого он ударяет молотком по различным сухожилиям, например, по пятке или под коленом, и проверяет реакцию.
Генетические исследования
Наиболее надежным методом выявления (наследственной) спинальной мышечной атрофии является генетический анализ. Врачи ищут доказательства измененного (мутировавшего) гена SMN1 и количества существующих копий SMN2.
Общее правило - диагностировать и лечить (наследственную) СМА как можно раньше. В зависимости от формы и доступного лечения, на двигательное развитие можно положительно повлиять до того, как мотонейроны спинного мозга будут необратимо повреждены.
Дальнейшие исследования в SMA
При подозрении на СМА врачи часто измеряют скорость проводимости нервов (электронейрография) и мышечную активность (электромиография). При необходимости также исследуют мышцы с помощью ультразвука (миосонография) или магнитно-резонансной томографии (МРТ).
Кроме того, врачи устраивают анализы крови. Если есть спинальная мышечная атрофия, некоторые параметры могут быть изменены: например, повышен уровень креатинкиназы (CK, типичный мышечный фермент).
Лечение мышечных атрофий позвоночника
Лечение спинальной мышечной атрофии комплексное. Долгое время причинная терапия ни при каких формах СМА была невозможна. Однако достижения в области медицинских исследований предоставляют врачам новые варианты лечения, которые существенно помогают людям с проксимальной СМА (дефект гена SMN на хромосоме 5).
Кроме того, врачи концентрируются на облегчении симптомов и предоставлении максимально возможной поддержки пострадавшим (например, физиотерапия, респираторная терапия, психотерапия, возможно, хирургическое вмешательство).
Лечебная терапия
Новые подходы к лечению пациентов, у которых СМА основана на известном дефекте гена SMN, вмешиваются непосредственно в сам генетический материал или в последующую обработку генетической информации.
Цель состоит в том, чтобы дать возможность телу пациента самостоятельно производить достаточное количество белка SMN, который имеет решающее значение для мотонейронов.
При спинальной мышечной атрофии доступны следующие варианты лечения:
- Модуляторы сплайсинга (Nusinersen, Risdiplam): эти препараты непосредственно вмешиваются в дальнейший процессинг молекул матричной РНК. Они усиливают те процессы, которые доставляют большее количество белка SMN из интактного гена SMN2.
- Генная заместительная терапия (онасемноген Abeparvovec): эта терапия воздействует непосредственно на геном человека. Неисправная копия гена SMN1 заменяется в пораженных клетках функциональной генной конструкцией, поставляемой извне.
Модуляторы сварки
В случае дефекта гена SMN1 организм может альтернативно продуцировать белок SMN из родственного гена SMN2. Замещающий ген SMN2 «вскакивает», но этого недостаточно. Причина: белки в SMN2 обычно слишком короткие и быстро расщепляются.
Это связано с процессингом соответствующей информационной РНК SMN2 (мРНК SMN2). Он передает информацию о конструкции из генома (ДНК) в места производства белка (рибосомы).
Для этого сначала считывается ген SMN2 в геноме. Предварительно продуцируется РНК-мессенджер SMN2. Помимо прочего, он должен быть подвергнут дальнейшей обработке посредством так называемого сращивания. Только тогда возникает зрелая информационная РНК. Специальные клеточные комплексы, рибосомы, затем читают зрелую информационную РНК и, таким образом, производят белок SMN2. И именно он укорачивается и нестабилен, быстро разбирается и не может взять на себя функцию SMN1.
Чтобы изменить это, активные ингредиенты Нусинерсен и Рисдиплам влияют на дальнейшую обработку предварительной матричной РНК. В результате эти так называемые модуляторы сплайсинга в конечном итоге увеличивают количество пригодных для использования белков SMN и, таким образом, могут обеспечить их достаточное количество.
Нусинерсен
Препарат Нусинерсен представляет собой так называемый «антисмысловой олигонуклеотид» (АСО). Он был одобрен Европейским агентством по лекарственным средствам в 2017 году. ASO - это искусственно созданные и специально адаптированные молекулы РНК. Они специфически и точно связываются с матричной РНК SMN2. Это предотвращает их дальнейшую некорректную обработку в клетке человека.
В частности: Нусинерсен предотвращает ошибочное вырезание важной информации (экзон 7) из матричной РНК SMN2. Расположение экзона 7 заставляет организм впоследствии производить более функциональный белок SMN.
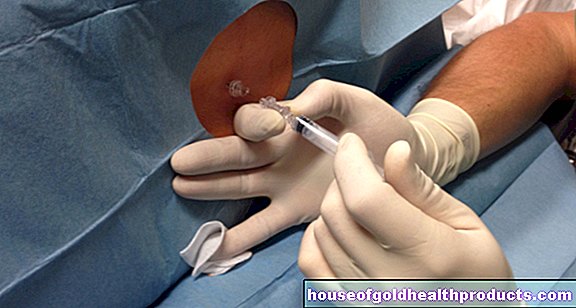
Нусинерсен вводится через так называемую люмбальную пункцию. Это означает, что препарат вводится в позвоночный канал с помощью шприца. Эта терапия повторяется через равные промежутки времени в несколько месяцев. В первый год лечения больные получают шесть, затем три дозы в год.
Пациенты обычно хорошо переносят препарат. Нусинерсен приводит к более благоприятному течению болезни. Исследования показали, что у многих пациентов улучшается подвижность: во многих случаях можно было свободно сидеть и самостоятельно поворачивать тело. Побочные эффекты и осложнения связаны, в том числе, с люмбальной пункцией (например, головная боль, инфекции мозговых оболочек).
Рисдиплам
Европейская комиссия одобрила Рисдиплам в качестве третьего препарата против 5q-ассоциированной СМА (типы 1-3 или от одной до четырех копий гена SMN2) в марте 2021 года. Рисдиплам принимают ежедневно в виде растворенного порошка. Точная доза рассчитывается исходя из возраста и массы тела.
В отличие от Нусинерсена, Рисдиплам - это не «антисмысловой олигонуклеотид», а небольшая молекула. Эта молекула связывается с информационной РНК для белков SMN2 и таким образом стабилизирует их. В результате создаются более функциональные белки SMN.
Общие побочные эффекты Рисдиплама включают дискомфорт в желудочно-кишечном тракте, сыпь, лихорадку и инфекции мочевыводящих путей.
Заместительная генная терапия
Другой подход к лечению проксимальной спинномозговой мышечной атрофии основан на так называемой генной заместительной терапии. Дефектный ген SMN1 - отправная точка прогрессирующей СМА - «заменяется» новой функциональной копией гена.
Действующий ингредиент Onasemnogen Abeparvovec (AVXS-101), работающий по этому принципу, получил условное разрешение на продажу для лечения малышей и детей от Европейского агентства по лекарственным средствам (EMA) в мае 2020 года.
Согласно информации EMA, препарат можно применять при СМА 1 типа. При всех других формах болезни SMA генетические характеристики (количество копий SMN2) решают, является ли генная заместительная терапия вариантом.
С помощью Онасемногена Abeparvovec функциональная копия гена SMN1 человека вводится в пораженные клетки спинного мозга и ствола головного мозга. Это делают определенные вирусы, которые служат «переносчиками» нового генетического материала - так называемые аденоассоциированные вирусные векторы (векторы AAV).
Конструкции векторных генов вводятся однократно в виде инфузии через вену в кровоток и оттуда распределяются по всему телу. Из-за еще не полностью сформированного гематоэнцефалического барьера у маленьких детей эти переносчики также могут попадать в ткани спинного мозга.
Предпочтительно связывая эти векторы со специальными поверхностными структурами моторных нейронов, они предпочтительно захватывают генетический материал, чтобы затем независимо продуцировать белок SMN.
Лечение может улучшить двигательные функции и привести к устойчивому успеху в развитии (например, сидение, ползание и ходьба без поддержки). Во время лечения иногда могут значительно увеличиваться показатели печени, но может уменьшаться количество тромбоцитов в крови. Также часто наблюдаются лихорадка и рвота. Чтобы уменьшить побочные эффекты, пациентам назначают кортикостероиды («кортизон») в течение нескольких недель.
Соответствующее возрасту моторное развитие, как правило, возможно только в том случае, если генная терапия началась бессимптомно. Лечение проходит в специализированных нервно-мышечных лечебных центрах.
физиотерапия
Физиотерапия продолжает оставаться важным элементом лечения СМА. Не все формы СМА можно лечить с помощью новых подходов к лечению. Регулярная лечебная физкультура предназначена для поддержания физических возможностей и замедления разрушения мышц.
Физиотерапевт пассивно перемещает части тела, которые уже парализованы. Последовательности активных движений, в свою очередь, тренируются для поддержки подвижности и силы мышц. Также могут помочь массаж или тепловые и холодные процедуры. Они также помогают расслабиться и, при определенных обстоятельствах, замедлить дальнейшее вырождение.
Логопедия
В некоторых случаях СМА поражает говорящие и глотательные мышцы. Тогда помогает логопедическое упражнение. Это побуждает детей учиться говорить. Даже у пожилых пациентов это обычно может замедлить ухудшение речи. Логопеды также тренируют правильное глотание.
И физиотерапевты, и логопеды поддерживают пострадавших с помощью целенаправленной дыхательной терапии.
Обезболивающее лечение
Обезболивание играет важную роль, особенно на более поздних стадиях заболевания. Врачи используют обезболивающие, чтобы уменьшить страдания пострадавших.
операция
Поскольку спинальная мышечная атрофия может привести к серьезному искривлению позвоночника (сколиозу), врачи иногда рассматривают возможность хирургического вмешательства. Тем самым они специально укрепляют позвоночник.
Это дает пострадавшим (определенную) дополнительную устойчивость туловища, которая не только позволяет принимать более прямую осанку, но также защищает кости и суставы. Операция на позвоночнике также может помочь при прогрессирующих проблемах с дыханием.
Психотерапевтическая помощь
Нервно-мышечные заболевания, такие как мышечная атрофия позвоночника, представляют собой серьезный психологический стресс. Пациенты и родственники обрабатывают диагноз на индивидуальных и групповых занятиях под руководством психотерапии и разрабатывают стратегии, позволяющие лучше справиться с этим заболеванием.
Группы самопомощи и представители пациентов также предлагают важную поддержку. Они информируют, консультируют и поддерживают пострадавших и их родственников, чтобы справиться с проблемами болезни СМА.
Шансы на выздоровление после мышечной атрофии позвоночника
Если есть мышечная атрофия позвоночника, прогноз зависит в первую очередь от соответствующей формы. Чем позже появятся симптомы, тем лучше течение. Кроме того, чем раньше врачи диагностируют спинальную мышечную атрофию, тем раньше они могут начать соответствующие лечебные мероприятия, даже до того, как мотонейроны будут необратимо повреждены.
Новые варианты лечения с помощью модуляторов сплайсинга и генной заместительной терапии обладают большим потенциалом в лечении проксимальной СМА, особенно при раннем начале лечения. Однако данные для надежного долгосрочного прогноза все еще не получены. Только дальнейшие исследования и тесные наблюдения за безопасностью лекарств могут обеспечить дополнительную уверенность в ближайшие (месяцы и) годы. С новыми лекарствами, по крайней мере, возможен долгосрочный контроль над болезнью или даже лечение.
Обычно серьезным заболеванием является СМА типа 1. Дети, у которых развивается СМА типа 1, имеют (без лечения) очень ограниченную продолжительность жизни. Быстро нарастающая мышечная слабость по всему телу также влияет на дыхание. Последствия - острая пневмония и даже дыхательная недостаточность. Больные дети умирают в течение первых нескольких лет жизни.
При СМА 2 типа прогноз немного лучше. Ожидаемая продолжительность жизни варьируется в зависимости от точной степени тяжести заболевания: некоторые умирают в детстве, но большинство из них достигают юношеского возраста. Рано или поздно - при желании - дыхание придется поддерживать в более тяжелых формах. Пострадавшие люди остаются мобильными с помощью инвалидной коляски.
При СМА 3 типа прогноз значительно лучше, особенно если первые симптомы появляются поздно. Производительность постепенно ухудшается в течение нескольких лет. В пожилом возрасте может потребоваться инвалидная коляска или даже постоянный уход. Продолжительность жизни практически не ограничивается мышечной атрофией позвоночника 3 типа.
Спинальная мышечная атрофия у взрослых (тип 4) протекает даже медленнее, чем тип 3. Люди обычно имеют нормальную продолжительность жизни.


.jpg)











.jpg)

