Синдром Ангельмана
Лиза Фогель изучала журналистику факультета с акцентом на медицину и бионауки в Университете Ансбаха и углубила свои журналистские знания, получив степень магистра в области мультимедийной информации и коммуникации. Затем последовала стажировка в редакции С сентября 2020 года она пишет в как независимый журналист.
Другие сообщения Лизы Фогель Весь контент проверяется медицинскими журналистами.Синдром Ангельмана (синдром счастливой марионетки) - редкое генетическое заболевание. Он проявляется, среди прочего, в умственных и физических ограничениях, нарушениях развития (особенно языковых) и гиперактивности. Что поражает, так это кукольный вид и счастливое выражение лиц пострадавших. Узнайте больше о редком заболевании!
Коды МКБ для этого заболевания: коды МКБ - это международно признанные коды медицинских диагнозов. Их можно найти, например, в письмах от врачей или в справках о нетрудоспособности. Q93

Краткий обзор
- Что такое синдром Ангельмана? Редкое генетическое заболевание, проявляющееся в умственных и физических ограничениях в развитии ребенка.
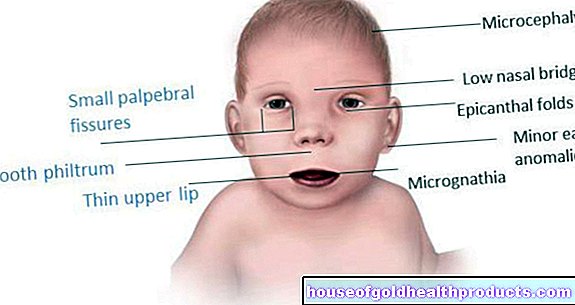
- Симптомы: кукольные черты лица, нарушение развития, нарушение координации, отсутствие или почти полное отсутствие языкового развития, снижение интеллекта, судороги, беспочвенный смех, приступы смеха, чрезмерное слюнотечение, радостное размахивание руками.
- Причины: генетический дефект на хромосоме 15
- Диагностика: в том числе беседа, физикальное обследование, генетические обследования.
- Терапия: нет причинно-следственной терапии; Поддерживающая, например физиотерапия, логопедия, трудотерапия; возможно, лекарства для облегчения симптомов (например, в случае судорог)
- Прогноз: нормальная продолжительность жизни; невозможна независимая жизнь
Синдром Ангельмана: определение
Синдром Ангельмана (СА) вызывается генетическим дефектом на хромосоме 15. Этот дефект нарушает физическое и умственное развитие пострадавших. Нарушения речевого развития, двигательная неуверенность и счастливое лицо - самые заметные симптомы синдрома Ангельмана.
Название «Синдром Ангельмана» происходит от первооткрывателя болезни, английского педиатра Гарри Ангельмана. В 1965 году он сравнил клинические фотографии трех детей с кукольными чертами лица. Дети много смеялись и делали резкие движения - как марионетки. Это привело к английскому названию «Happy Puppet Syndrome» (счастливая марионетка).
Синдром Ангельмана встречается у обоих полов. Риск заболевания составляет примерно 1:20 000. Это делает синдром редким заболеванием.
Синдром Ангельмана: симптомы
При рождении дети с синдромом Ангельмана нормальны. Расстройства моторного и когнитивного развития становятся все более заметными только в младенчестве и раннем детстве. Особенности генетического заболевания:
- задержка моторного развития
- нарушение координации
- часто нет или почти нет языкового развития
- снижение интеллекта
- гиперактивное, резвое поведение
- беспочвенный смех
- Приступы смеха
- радостные жесты (например, размахивание руками)
- чрезмерное слюнотечение
- частое высовывание языка
У некоторых детей с синдромом Ангельмана также есть:
- Микроцефалия (аномально маленькая голова) - не при рождении, а по мере развития
- Судороги
- Изменения электрической активности мозга
- очень светлая кожа и глаза из-за снижения пигментации (гипопигментация)
- Косоглазие (косоглазие)
Синдром Ангельмана: причины
Причина синдрома Ангельмана - генетический дефект на хромосоме 15: у больных нарушена функция гена UBE3A. Этот ген обычно производит фермент, который участвует в расщеплении поврежденных или избыточных белков в клетках. Таким образом, это помогает клетке нормально работать.
Ген UBE3A расположен в районе хромосомы 15q11q13. Там гены подвергаются так называемому «геномному импринтингу». Это означает, что они активны только на одной из родительских хромосом (в клетках нашего тела есть две копии каждой хромосомы - одна от матери и одна от отца). Это регулируется химическим процессом - метилированием: метильные группы, присоединенные в определенных точках, могут включать или выключать ген.
Ген активен на обеих хромосомах во многих клетках организма, но не в нервных клетках мозга: у многих людей ген UBE3A на отцовской хромосоме 15 отключается путем импринтинга. В результате UBE3A активен только на материнской хромосоме 15 в головном мозге. Это также означает: если копия материнского гена показывает ошибку, это не может быть компенсировано неиспользованной копией родительского гена. И именно такая комбинация возникает при синдроме Ангельмана: сегмент отцовского гена выключен, сегмент материнского гена неисправен.
Основная генетическая ошибка может быть разных типов:
- Делеция: около 75 процентов всех людей с синдромом Ангельмана не имеют соответствующей области 15q11q13 с геном UBE3A на материнской хромосоме 15. Поскольку соответствующая область на отцовской хромосоме 15 «выключается» импринтингом, организм может использовать фермент, план строительства которого сохраняется в Gen UBE3A, не создавайте.
- Мутация в гене UBE3A: спонтанно происходящее изменение в гене приводит к потере содержащейся в нем информации. Это верно от пяти до десяти процентов людей с синдромом Ангельмана. Примерно в каждом пятом случае происходит семейная мутация: здесь мать уже несет генетическое изменение в хромосоме своего отца.
- Две отцовские хромосомы 15: пострадавший унаследовал обе хромосомы 15 от своего отца, ни одну от матери (с медицинской точки зрения это называется «отцовская однопородная дисомия 15»). Итак, активного гена UBE3A нет. Это относится примерно к одному-двум процентам всех пациентов с синдромом Ангельмана.
- Ошибка импринтинга: ген UBE3A на материнской хромосоме 15, как и ген на отцовской хромосоме 15, отключается импринтингом. Кроме того, определенная часть хромосомы может отсутствовать (делеция). Ошибка импринтинга обнаруживается от одного до четырех процентов случаев синдрома Ангельмана.
В остальных 10-15% случаев причина синдрома Ангельмана неизвестна. Кстати: если материнский ген выключен, а отцовский ген неисправен, дети страдают так называемым синдромом Прадера-Вилли.
Синдром Ангельмана наследственный?
В целом риск рецидива синдрома Ангельмана невысок. Это означает риск того, что у родителей пораженного ребенка будут другие дети, у которых также есть синдром. Однако в отдельных случаях этот риск во многом зависит от генетического дефекта, лежащего в основе синдрома Ангельмана.
Например, при синдроме Ангельмана из-за двух отцовских хромосом 15 (отцовская однопородная дисомия 15) он составляет менее одного процента. С другой стороны, синдром Ангельмана из-за ошибки импринтинга с потерей определенного сегмента гена (делеция IC) может возникать в половине всех случаев у братьев и сестер.
Этот повышенный риск также существует при мутации UBE3A - при условии, что мать уже является носителем генетического дефекта (примерно в 20% всех случаев мутации). В таких случаях мать унаследовала мутацию от отца. Таким образом, UBE3A изменяется в отцовской хромосоме матери. Если это выключено, мать не заметит мутации. Однако она может передать хромосому своим детям, где она - тогда как материнская хромосома - может вызвать синдром Ангельмана.
Теоретически пациенты с синдромом Ангельмана могут воспроизводить потомство. В зависимости от того, когда произошли причинные хромосомные изменения (например, уже во время развития зародышевых клеток или сразу после оплодотворения), иногда очень высок (до 100 процентов) риск того, что пострадавшие передадут болезнь. Однако достоверных данных об этом нет. В сентябре 1999 года был единичный случай: здесь передала болезнь мать, страдавшая синдромом Ангельмана.
Синдром Ангельмана: диагностика
Если вы заметили у своего ребенка описанные выше симптомы, в первую очередь обратитесь к педиатру. Он может более точно сузить возможные причины и при необходимости направить вас и вашего ребенка к специалисту.
анамнез
Первым шагом в диагностике является тщательный сбор анамнеза. Врач задаст вам различные вопросы о вашем ребенке, например:
- Какие изменения вы заметили в своем ребенке?
- Есть ли у вашего ребенка какие-либо жалобы на физическое состояние?
- Ваш ребенок может сидеть?
- Ваш ребенок тянется к предметам?
- Ваш ребенок говорит?
- Ваш ребенок часто бывает заметно весел или взволнован?
- Ваш ребенок смеется в неподходящих ситуациях, например, когда ему больно?
Физическое обследование
Затем следует физический осмотр. Педиатр проверяет, насколько регулярно у ребенка развиваются двигательные и умственные навыки. Для этого используются простые упражнения: например, ребенок должен сосредоточиться на игрушках или выборочно дотянуться до строительного блока. Также врач обращает внимание на мимику ребенка. Частый смех, кукольные черты лица и слюни - все это признаки синдрома Ангельмана.
Если после медицинского осмотра появится подозрение на редкое заболевание, врач направит вас к неврологу и генетику.
Генетические тесты
Генетическое тестирование - важная часть диагностики синдрома Ангельмана. Врачу нужен небольшой образец клеток ребенка, который он может получить из слизистой оболочки полости рта, например, взяв образец крови или взяв мазок. Затем генетический материал (ДНК) этих клеток или соответствующего участка хромосомы 15q11q13 исследуется более подробно в лаборатории.
На первом этапе врачи обращают внимание на характер метилирования хромосомного сегмента (анализ / тест метилирования). Дальнейшие тесты на тех же образцах (анализ делеций, анализ мутаций) помогают более точно определить причину синдрома Ангельмана. Для этого также может потребоваться изучить генетический состав родителей. Таким образом, врачи могут определить, есть ли уже генетический дефект.
Дальнейшие исследования
Часто бывает полезно дальнейшее обследование. Например, ЭЭГ можно использовать для обнаружения изменений в электрической активности мозга, как это часто бывает при синдроме Ангельмана. Также могут быть показаны офтальмологические обследования.
Синдром Ангельмана: терапия
Синдром Ангельмана неизлечим - невозможно устранить генетическую причину заболевания. Однако раннее вмешательство может положительно повлиять на двигательное и умственное развитие пострадавших. Например, может помочь регулярная физиотерапия. Он может улучшить моторику детей, уменьшить ограниченную подвижность и помочь предотвратить вторичные заболевания, такие как деформации позвоночника. Детям с синдромом Ангельмана также полезны другие методы терапии, такие как трудотерапия и логопедия.
Кроме того, некоторые симптомы и состояния, связанные с синдромом Ангельмана, могут потребовать целенаправленного лечения. Например, противосудорожные препараты (противоэпилептические препараты) помогают от судорог, а транквилизаторы (седативные средства) помогают при тяжелых нарушениях сна.
На веб-сайте Verein Angelman e.V. вы найдете много информации о синдроме Ангельмана, отчеты об опыте и событиях для пострадавших, а также контактных лиц для пострадавших во всех регионах Германии.
Синдром Ангельмана: течение болезни и прогноз
Первый год жизни
Младенцы с синдромом Ангельмана чаще испытывают проблемы с грудным вскармливанием, сосанием и глотанием. Они часто высовывают язык или пускают слюни. Кроме того, дети с синдромом Ангельмана часто плюются (что часто принимают за непереносимость пищи или рефлюксную болезнь). Частая рвота может привести к опасной потере веса.
В возрасте от трех до шести месяцев дети с синдромом Ангельмана часто начинают улыбаться. Они много хихикают и булькают и часто высовывают языки во время этих вспышек радости.
Задержка моторного развития обычно заметна в возрасте от 6 до 12 месяцев: дети не ползают и не сидят. Движения верхней части тела часто шаткие. Это, в свою очередь, затрудняет сидение.
У части пострадавших уже случаются судороги в возрасте 12 месяцев.
От одного до трех лет
В течение первых трех лет жизни нарушение развития при синдроме Ангельмана становится очень очевидным. Дети больше страдают легкими припадками. Некоторые из них гиперактивны, перевозбуждены и постоянно находятся в движении. Многие имеют тенденцию все время засовывать руки или игрушки в рот или часто высовывать язык и пускать слюни. Если дети особенно возбуждены, они часто будут чрезмерно смеяться и махать руками с вытянутыми руками.
Нарушение языкового развития впервые проявляется в этом возрасте. Дети «бормочут» про себя или кричат и пищат, однако могут произнести только несколько легко понятных слов или вообще не произносить их и обычно используют их без контекста.Но они понимают язык, и есть также социальное взаимодействие с другими людьми.
Половое созревание и взрослая жизнь
У детей с синдромом Ангельмана половое созревание часто наступает на 3-5 лет позже. Однако затем половая зрелость развивается нормально. По-прежнему отсутствует языковое развитие, если понимание языка часто присутствует. Приступы во взрослом возрасте обычно хорошо купируются лекарствами.
Люди с синдромом Ангельмана имеют нормальную продолжительность жизни. Самостоятельная жизнь невозможна из-за умственных ограничений.
теги: малыш желание иметь детей здоровье мужчины
















.jpg)